Biography
My research project aims to study the role played by long non-coding RNAs (lncRNAs) in the adaptation of the root of plants to the environment, via multiscale quantitative approaches. As a plant molecular biologist, I am interested in the regulation of genes expression by these lncRNAs, that will impact root system architecture (RSA).
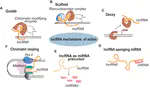
Over the past decades, advances in RNA sequencing methods (RNA-Seq) have revealed that a large proportion of eukaryotic genomes are transcribed outside the coding regions. These non-coding RNAs (ncRNAs) have been shown to play an emerging role in the regulation of gene expression and could be then good candidates for the fine control of the adaptation to the environment.
Learn more about my research by browsing the projects I am involved in.
- long non-coding RNAs (lncRNAs)
- root growth
- quantitative regulations
-
PhD in Plant Science, 2008
Université Paris-Sud, France
-
Master of Science, specialization in Plant Science, 2005
Université Paris-Sud, France
-
Agronomic ‘Engineer’ degree, 2005
INA P-G (today AgroParisTech), France
Projects

Featured Publications



Experience
Quantitative control of root architecture by non-coding RNAs.
Study of the role of long non-coding RNAs using multi-scale approaches (from the scale of the gene to the organ). In particular their role in the regulation of gene expression and their influence on the architecture of the root system.
Role of non-coding RNAs in adaptation to the environment.
Study of the importance of non-coding RNAs in the adaptation of Arabidopsis accessions to phosphate deficiency: sequence conservation, expression variation and control of root architecture.
The growth of Arabidopsis leaf at the cellular level.
Reconstruction of a 3D model of leaf primordia at the cellular scale to identify cellular characteristics associated with tooth formation.
Modeling the growth of Arabidopsis root
Reconstruction of a 3D model of the root at the cellular level from confocal images. Characterization at the cellular level of the influence of light on root growth.
The role of NAM / CUC genes in leaf development
Determination of the expression of NAM / CUC genes in different compound leaf species by in situ hybridization. Functional analysis of the role of these genes via mutants, transformants and silenced plants by VIGS. Characterization of the genetic interactions between the CUC2 / miR164A genes and other factors involved in Arabidopsis thaliana leaf serration.