Whole-genome landscape of Medicago truncatula symbiotic genes.
Abstract
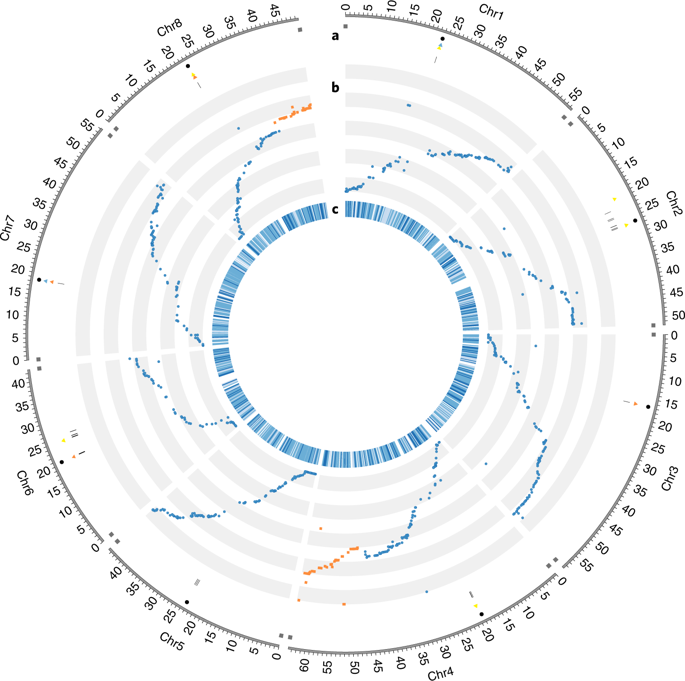
Advances in deciphering the functional architecture of eukaryotic genomes have been facilitated by recent breakthroughs in sequencing technologies, enabling a more comprehensive representation of genes and repeat elements in genome sequence assemblies, as well as more sensitive and tissue-specific analyses of gene expression. Here we show that PacBio sequencing has led to a substantially improved genome assembly of Medicago truncatula A17, a legume model species notable for endosymbiosis studies , and has enabled the identification of genome rearrangements between genotypes at a near-base-pair resolution. Annotation of the new M. truncatula genome sequence has allowed for a thorough analysis of transposable elements and their dynamics, as well as the identification of new players involved in symbiotic nodule development, in particular 1,037 upregulated long non-coding RNAs (lncRNAs). We have also discovered that a substantial proportion ( 35% and 38%, respectively) of the genes upregulated in nodules or expressed in the nodule differentiation zone colocalize in genomic clusters (270 and 211, respectively), here termed symbiotic islands. These islands contain numerous expressed lncRNA genes and display differentially both DNA methylation and histone marks. Epigenetic regulations and lncRNAs are therefore attractive candidate elements for the orchestration of symbiotic gene expression in the M. truncatula genome.